What is CDH?
Congenital diaphragmatic hernia (CDH) is a birth defect that occurs in about 1/2500 babies and that consists of a hole in the diaphragm (Figure 1). The diaphragm is the thin muscle separating the chest from the belly. Normally, the lungs and heart are situated above the diaphragm in the chest and the stomach, the bowels and the liver below in the abdomen. When there is a hole in the diaphragm in over 80% of cases, the hole is situated on the left side of the diaphragm; in nearly 20% it is on the right side and in exceptional cases it involves both sides. The defect in the muscle appears as early as 8 weeks of gestation (normal duration of pregnancy is 40 weeks) and the reason why it appears is most often unknown. Through the defect in the diaphragm the intestines, the stomach and in some cases the liver or even the spleen can ascend to the chest. This limits the space for lung growth during pregnancy.►
CDH may occur as an isolated problem or in association with other problems such as malformations from other organs, abnormal chromosomes or a syndrome, which is a group of signs and symptoms that occur together and characterize a particular condition. Babies with combined problems usually face a rather poor prognosis. However, in the majority of cases the defect is the only problem (this is what we call ‘isolated’ CDH) and these infants have a much better prognosis.
Diaphragmatic hernia can also appear later in life and even in adults. Depending on the symptoms the defect needs surgical repair, which for adults is all in all a very reasonable operation. Comparable surgery can be done in infants with the disease. However the real problem for babies with CDH does not lie in the repair of the muscular defect, but in the problems the defect may already have caused before birth by compressing the developing lungs.►Indeed, from birth on, the baby needs its lungs for breathing and oxygen uptake. Because of the impaired lung development these newborns face different respiratory problems, which need to be solved before any attempt for surgery is done.
Consequences of CDH
The consequences of CDH can be separated in 2 main categories:
Consequences of CDH before and at birth
The most important consequence of the diaphragmatic defect is the impaired lung development during pregnancy. CDH-babies have smaller lungs, especially on the side of the defect. This underdevelopment of the lungs is called “pulmonary hypoplasia”. These lungs are also less elastic, have less airways and surface for oxygen uptake and less and thicker blood vessels.
During the baby’s stay in the womb, the small lungs do not pose a problem, since the fetus does not obtain its oxygen through its lungs. During pregnancy, it gets all its oxygen and nutrients from its mother through the placenta. Only 1% of babies with CDH develops problems before birth and dies in the womb for unknown reasons.
A more frequently seen problem during pregnancy is that the baby swallows less easily because the stomach and intestines are displaced in the thorax. This can then lead to an increased volume of amniotic fluid.
Babies with CDH typically face the consequences of the defect only at birth, when they need their own lungs for oxygen uptake and carbon dioxide elimination. It is also for this reason that it is important that the diagnosis of CDH is made before birth. If doctors are aware that the baby has a problem with its lungs, they can organize its birth in optimal conditions. This means that they can plan it at a center that is fully equipped, staffed and experienced to handle such a difficult start.
At birth, the baby’s lungs can be that small that the baby is unable to transport enough oxygen into its blood, or carbon dioxide out of the blood. The baby may also have a high blood pressure within its lungs because its blood vessels are fewer in number and more stiff.
This is why doctors will assist the baby with its breathing using modern tools. Basically these consist of ventilation machines that help or replace the lungs, drugs that decrease the blood pressure in the lungs and in some cases a heart-lung machine.
Once the baby is sufficiently stabilized from a respiratory point of view, the diaphragmatic defect is surgically repaired. This is usually done within the first 10 days of the baby’s life and represents another critical moment for the neonate. The repair itself can either be done by suturing the 2 borders of the remaining diaphragm together or by sewing a small tissue (Gore-Tex) patch in the defect. After successful repair, there is more space for the lungs in the chest, which then can catch up growth to a certain degree. Of course, after the operation, the baby still needs time to recover in the hospital before he/she can go home.
The degree of severity of the problems at birth depends on the size of the lungs and therefore is different from case to case. Overall, the smaller the lungs, the more difficult the baby’s start of life will be. Worldwide, up to 30 % of babies with isolated CDH do not survive the early weeks of life. Part of this is because the diagnosis was not made before birth and/or birth took place in unfavorable conditions. But also in very experienced centers and despite all efforts, some babies just have lungs that are so small that every therapy that can be offered after birth falls short. The number of babies that survive when born in optimal conditions may vary from place to place and depends on factors such as experience and used technologies as well as on handling of complications.
Consequences of CDH in later life
Later in life, most grown up children will not have major lasting problems. Obviously there is the scar from the surgical repair of the defect. Also a number of infants remain limited to a certain degree in their capacity to perform physical activities, mainly because their lungs are still smaller than those of a normal child. Some babies may also experience problems with feeding and as a consequence problems with thriving as the connection between the esophagus and the stomach may not function normally, leading to reflux. Most babies initially require help with feeding and it takes time before they can swallow without help. The baby may also require a medical treatment for its reflux or, in more difficult cases, it may benefit from an operation that decreases the inlet from the esophagus into the stomach.
Another less common chronic health problem is scoliosis (this is a deformity in the spine), because the thorax does not grow equally on the side of the defect. Also, longer lasting respiratory problems such as asthma or chronic airway infections are more frequently in CDH infants. All of these problems can be treated if necessary. A minority of children may have major long-term problems of which the most important is prolonged respiratory restriction or even oxygen dependency. For an unknown reason, hearing problems can occur in infants with CDH.
Referral for specialized care prior to birth
CDH is typically diagnosed on a routine ultrasound scan during pregnancy. This diagnosis obviously creates a lot of anxiety with parents, who therefore should talk to doctors with experience in taking care of these babies. These specialists can look back on a wide experience with many babies with a similar condition and should be able to answer the parent’s questions and decrease their anxiety.
Parents usually have concerns on immediate and remote consequences of the disease for their baby and their family. Fortunately, a lot of these questions can already be answered, at least partly, before birth.
Prenatal diagnosis
Just as the baby will need specialized care during pregnancy and after birth, the parents need specialized counseling as well; preferentially before birth. For that reason they should be referred to a tertiary care center. In many countries this is limited to a few centers, prompting some travelling.
At such a center the pregnancy is re-evaluated by fetal medicine specialists to determine as much as possible whether CDH is an isolated problem or whether it is associated with any other problems. The workup will be comprehensive, including a detailed ultrasound of the entire baby, an examination of the fetal heart and sampling of the amniotic fluid to examine the genetic material or ‘chromosomes’. Moreover, the lungs will be measured to assess their size and advanced examinations like fetal Magnetic Resonance Imaging (MRI) or 3-dimensional ultrasound may provide additional information.
After these examinations the parents get the opportunity to talk with the different specialists who will take care of their baby. These can be, next to the fetal medicine specialists (who look after the unborn baby), an obstetrician (who takes care of the mother during the pregnancy and who does the delivery), a neonatologist (who will take care of the baby immediately after birth) and a pediatric surgeon (who will repair the defect). This team will be counseling the parents based on the individual findings of all above mentioned examinations. No adequate estimation of the prognosis of the disease and the implications of CDH for any particular baby can be made prior to this stage and one must refrain from drawing early conclusions prior to this moment.
The rest of the pregnancy usually takes a normal course, but this must be followed closely and the moment of birth must be well timed such that maximal support can be offered to the baby. Moreover, the remaining months of pregnancy are often a period of anxiety and emerging questions for the parents. Therefore, regular consultation with the fetal medicine specialists, social workers, parents’ organizations or a psychologist may play an important role during the further pregnancy.
Prenatal evaluation of the prognosis
Today, fetal medicine specialists can already attempt to answer some of the questions from the parents before birth. They can estimate the prognosis of the baby in its individual situation rather than just citing the general statistics as mentioned above.
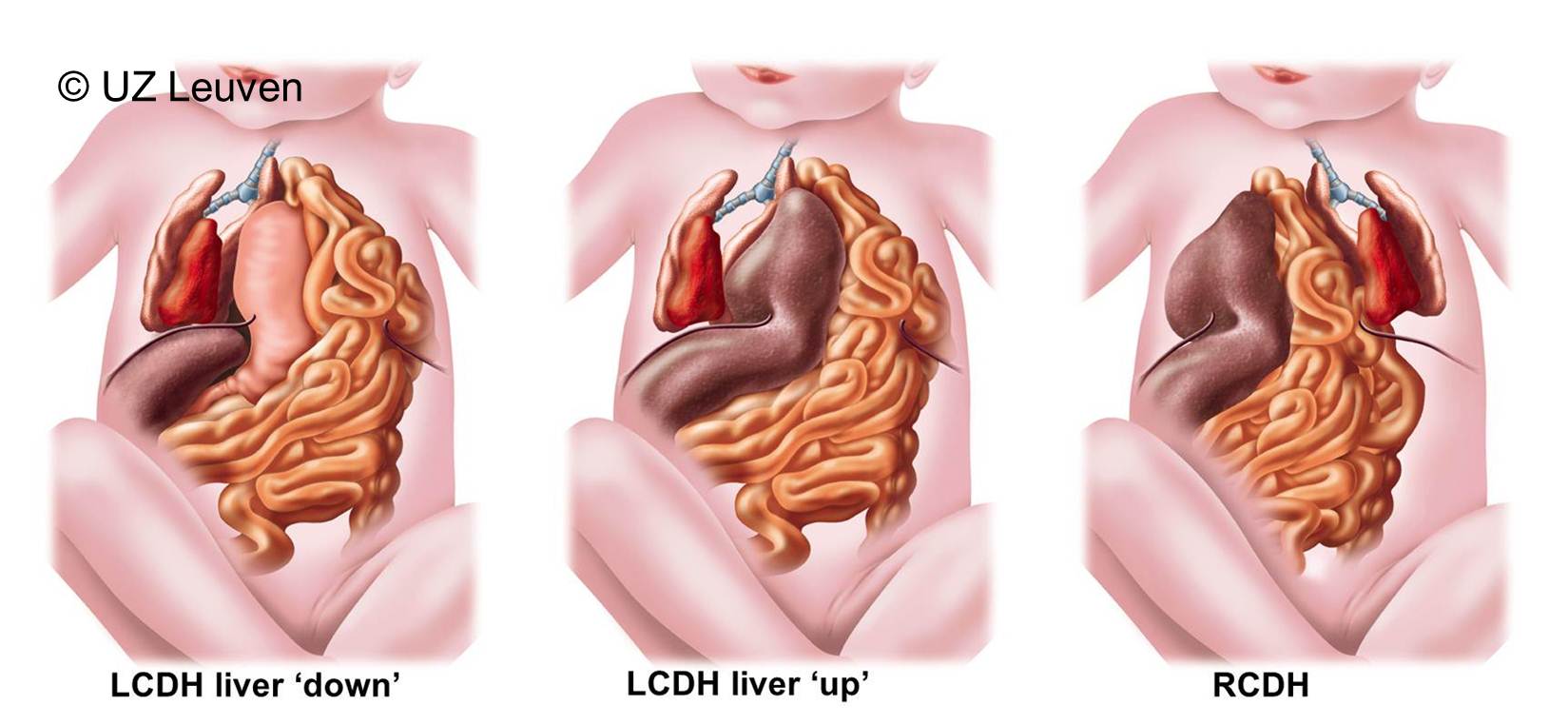
Indeed chances for survival can reasonably well be estimated by prenatal examination. The best known factors to predict survival are the position of the fetal liver and the size of the fetal lungs. Usually these 2 factors are assessed by ultrasound, but MRI may also be helpful.
The fetal medicine specialists will measure lung size, expressed as a lung-to-head ratio (LHR; Figure 2). This LHR is obtained by measuring the area occupied by the largest lung in the chest and dividing it by the head circumference. The LHR of the individual baby with CDH can then be compared to the LHR of normal babies. The doctor will finally express the LHR as a percentage of the normal. The lower this percentage, the smaller the lungs and the poorer the chances of survival. Next, the fetal medicine specialists will determine the position of the liver (Figure 3). The liver is either ascended or “up” (herniated) into the chest; if not it is “down” as in babies without CDH. When the liver is herniated, the defect is usually larger and therefore the disease is often more severe. Both factors (LHR and liver position) can be used together to predict the outcome of the individual baby, based on statistics gathered in several major centers over Europe that have gathered a large experience with CDH babies. The numbers displayed in the graph are therefore representative of what can be offered in optimal conditions in these centers.(Graph)
Based on these statistics, we therefore subdivide the fetuses in the following groups:
· Severe lung hypoplasia: the lung is that small that the vast majority of babies will die despite intensive care.
· Moderate lung hypoplasia: Between 40-60% of babies survive. A number of them will have ongoing problems. In about 30% there are breathing problems requiring oxygen therapy for at least one month after birth and in a few cases these are longer term.
· Mild lung hypoplasia: lung development is such that more that 60 to 95% of the babies will survive. The number of babies with persisting breathing problems in this group is minimal.
The size of the lung before birth and the fetal liver position also determine how long the baby will need ventilation or oxygen supplementation, how extensive the postnatal surgery will be, and the time the baby will need before it can be fed without medical assistance. Since all of this has been discovered only recently, the relationship between these measurements and how these babies do as teenagers, is yet unknown. There are ongoing efforts to make prediction more accurate, using newer imaging techniques, such as fetal MRI, 3D ultrasound, blood flow studies of the lung, reaction to oxygen, etc… At this time, all these techniques are still investigational and results of this research will be communicated as work progresses.
Planning of birth
Again, birth should be planned to take place in optimal conditions. This means that the baby should be born in a center that offers intensive neonatal care, has a large experience with CDH and has the necessary technical equipment and staff to provide good medical care. Moreover, birth should take place at a moment that medical and nursing staff is optimally present and prepared for the immediate neonatal management of the baby. Therefore, births of CDH babies are planned as much as possible. If there are no concurrent problems in pregnancy the doctors will want to plan delivery at a moment that the lungs of the baby are mature. In most centers this is around 38 weeks of gestation. Whether birth takes place by vaginal route or by caesarean section is a matter of planning and local habits, as neither mode of delivery has been shown to be superior over the other, as long as neonatal care-takers are optimally prepared.
Figure 1: Left congenital diaphragmatic hernia
Figure 2: LHR
Figure 3: CDH overview
 back
back